
Abstract
Programmable and modular systems capable of orthogonal genomic and transcriptomic perturbations are crucial for biological research and treating human genetic diseases. Here, we present the minimal versatile genetic perturbation technology (mvGPT), a flexible toolkit designed for simultaneous and orthogonal gene editing, activation, and repression in human cells. The mvGPT combines an engineered compact prime editor (PE), a fusion activator MS2–p65–HSF1 (MPH), and a drive-and-process multiplex array that produces RNAs tailored to different types of genetic perturbation. mvGPT can precisely edit human genome via PE coupled with a prime editing guide RNA and a nicking guide RNA, activate endogenous gene expression using PE with a truncated single guide RNA containing MPH-recruiting MS2 aptamers, and silence endogenous gene expression via RNA interference with a short-hairpin RNA. We showcase the versatility of mvGPT by simultaneously correcting a c.3207C>A mutation in the ATP7B gene linked to Wilson’s disease, upregulating the PDX1 gene expression to potentially treat Type I diabetes, and suppressing the TTR gene to manage transthyretin amyloidosis. In addition to plasmid delivery, we successfully utilize various methods to deliver the mvGPT payload, demonstrating its potential for future in vivo applications.
Similar content being viewed by others

Introduction
A compact, robust, and generalizable genetic perturbation system capable of precise genome editing alongside simultaneous and orthogonal modulation of endogenous gene expression in living cells is critical for therapeutic interventions of complex diseases, genetic screening, and metabolic engineering1,2,3. While CRISPR/Cas nucleases combined with transcriptional activators can achieve concurrent gene knockout and activation4,5, precise gene editing using Cas nucleases relies on homology-directed repair stimulated by double-stranded breaks (DSBs), which is inefficient in most therapeutically relevant cell types6,7,8,9. Further, DSBs often lead to unintended genetic alterations and cytotoxicity, posing safety concerns for therapeutic applications6,7,8,9. Base editors (BEs) provide an alternative approach, converting one nucleobase to another without causing DSBs10,11,12,13. However, BEs are limited to point mutations and lack orthogonality for multiplexed gene editing and activation14.
Prime editors (PEs) employ a nickase Cas9 (nCas9)-reverse transcriptase (RT) fusion protein and a prime editing guide RNA (pegRNA) to reverse transcribe a short RNA template into targeted DNA sites without requiring DSBs15,16,17. PEs enable a variety of precise gene editing capabilities, including base substitutions, insertions, deletions, and gene inversions and translocations15,16. Thus, we developed a PE-based multiplexed genetic perturbation technology, enabling simultaneous gene editing and transcriptomic modulation. For potent gene activation, we integrate PEs with the synergistic activation mediator (SAM) system18 using engineered MS2-binding stem-loops in the single guide RNA (sgRNA) to recruit the transcriptional fusion activator MS2–p65–HSF1 (MPH). For gene silencing, we use short-hairpin RNAs (shRNAs) that competitively hybridize with mRNA targets19, which is independent of gene editing and/or PE-SAM gene activation. Hence, concurrent use of an engineered compact PE, a recruitable transcriptional fusion activator, and a shRNA enables simultaneous and orthogonal gene editing, activation, and repression.
Here, we describe the minimal versatile genetic perturbation technology (mvGPT) built on our previously developed drive-and-process (DAP) array1,20, a compact and multiplexed RNA expression system for base and prime editing. The DAP array uses a 75 bp human cysteine tRNA (hCtRNA) as a promoter and spacer between RNA elements (Fig. 1a). Following endogenous hCtRNA processing, individual RNA subunits are released from the array, thus avoiding cumbersome individual promoters while retaining similar levels of RNA expression. When integrated into mvGPT, the DAP array orchestrates the production of RNAs with distinct functionalities for endogenous gene editing, activation, and repression at independent genomic loci. For gene editing, mvGPT utilizes pegRNA and nicking guide RNA (ngRNA) to direct our engineered compact PEs to the target loci, efficiently introducing precise genomic modifications. For gene activation, a truncated sgRNA containing MS2-binding stem loops recruits the MPH activation complex to PE, upregulating gene transcription. Finally, the DAP array-generated shRNA ensures potent gene repression through RNA interference (RNAi). We demonstrate mvGPT’s capacity for simultaneous and orthogonal genetic interventions by correcting the Wilson’s disease-related c.3207C>A mutation in the ATP7B gene, upregulating the PDX1 gene for treating Type I diabetes, and repressing the TTR gene to manage transthyretin amyloidosis. Moreover, we successfully deliver the mvGPT using mRNA, AAV, and lentivirus, highlighting its broad compatibility with different delivery systems.
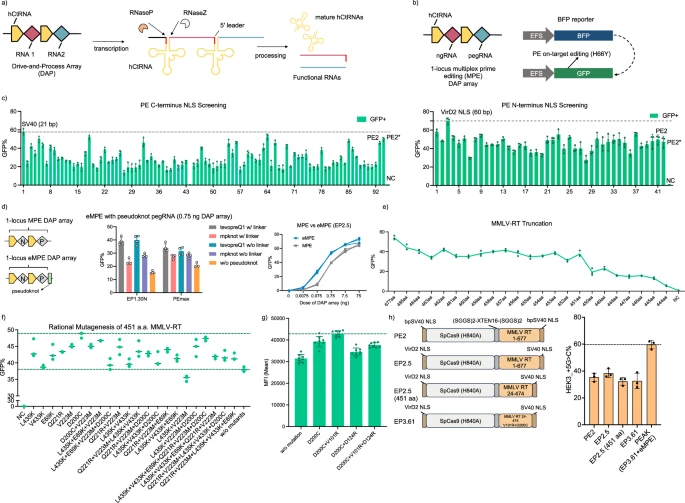
a Schematic of the drive-and-process (DAP) array. b Schematic of the BFP fluorescent reporter. On-target 1-locus MPE converts BFP to GFP and can be used as an indicator of prime editing efficiency. EFS, elongation factor 1α short promoter. c Evaluation of PE variants with different C-terminal (left) and N-terminal NLSs using the BFP-to-GFP reporter. Dashed lines show the highest GFP conversion achieved by the best variants: C-terminal SV40 and N-terminal VirD2. X-axes lists the PE variants tested. n = 3 biological replicates d. Integration of engineered pegRNA (epegRNA) into the DAP array. n = 4 (left) and n = 3 (right) biological replicates e Performance of PE variants with truncated MMLV-RT, as indicated by BFP-to-GFP conversion rates. n = 3 biological replicates. f Rational engineering of 451 aa MMLV-RT by introducing previously reported beneficial mutations. Upper dash line indicates the D200C mutant and lower dash line indicates the 451 aa MMLV-RT without additional mutations. n = 3 biological replicates. g Comparison among top-performing 451 aa MMLV-RT variants. n = 8 biological replicates. h Comparison of engineered prime editors and PE2 targeting the endogenous HEK3 locus in HEK293T cell. n = 3 biological replicates. Bars represent the mean ± S.D. for all plots. NC represents a non-transfected control for all relevant plots. Source data are provided as a Source Data file.
Results
Engineering a compact and efficient prime editing system
Before exploring PEs for gene regulation, we optimized the efficiency and compactness of the prime editing system. To enable high-throughput screening of engineered PE variants, we developed a reporter assay linking PE activity to the conversion of blue fluorescent protein (BFP) to green fluorescent protein (GFP), achieved by a C-to-T substitution that converts His to Tyr within the BFP Thr-His-Gly chromophore21 (Fig. 1b). We then screened 13 DAP arrays each containing different ngRNA/pegRNA pairs and found that the DAP array EP 1.11 demonstrated the highest prime editing efficiency, measured by the BFP-to-GFP conversion rates (Supplementary Fig. 1a–n). We subsequently integrated the EP1.11 DAP array and the BFP gene into the HEK293T genome via lentiviral infection, creating the BFP reporter v2 stable cell line to evaluate PE activity (Supplementary Fig. 2). Additionally, we developed a 3-color reporter system with an efficient 3-loci multiplex prime editing (MPE) DAP array to simultaneously report PEs’ ability to insert a 9-bp fragment to recover the EGFP TYG chromophore, delete a 6-bp pre-installed stop codons on mCherry, and substitute a 6-bp fragment to recover the TagBFP LYG chromophore (Supplementary Fig. 3a and supplementary Figs. 4, 5).
Building on the foundational PE2 system, we first aimed to improve its prime editing efficiency by optimizing the nuclear trafficking via nuclear localization signal (NLS) engineering22,23,24. We constructed 91 PE2 variants with different C-terminal NLSs and 31 variants with different N-terminal NLSs sourced from the NLSdb database25, followed by screening using the BFP reporter v2 stable cell line (Fig. 1c, Supplementary Fig. 6a). Substantial improvements were observed with the N-terminal VirD2 NLS or the C-terminal SV40 NLS (Fig. 1c). Combining these two best NLSs into one variant (EP2.5) produced a synergistic effect, resulting in a 7% increase in BFP-to-GFP conversion rates compared to PE2 with a N and C terminal BPSV40 NLSs (Supplementary Fig. 6b). This improvement was consistent across various genetic modifications when tested with the 3-color reporter cell lines, with EP2.5 showing a 10–18% increase in insertion efficiency, 9–14% increase in substitution efficiency, and 12–35% increase in deletion efficiency compared to PE2 (Supplementary Fig. 3d–f). To further improve prime editing efficiencies, we incorporated previously reported engineered pegRNAs (epegRNAs) into the DAP array, which each have structured 3’ motif for enhanced stability and resistance to degradation26 (hereafter referred to as eMPE) (Fig. 1d). eMPE consistently outperformed MPE across multiple PE variants and different DAP array dosages (Fig. 1d, Supplementary Fig. 7). Notably, the DAP array with the trimmed pseudoknot evopreQ1 (tevopreQ1, with linker) showed a 10–35% increase in prime editing efficiency, reflected by the BFP-to-GFP reporter, and was used for all further eMPE experiments (Fig. 1d).
Next, we truncated the Moloney Murine Leukemia Virus reverse transcriptase (MMLV-RT) used in PE2, guided by studies indicating that the RNase H domain and the first 23 amino acid (aa) residues of the MMLV-RT are non-essential27,28,29,30,31. Compared to the canonical PE2 with full length 677 aa (1–677) MMLV-RT, a truncated variant with only the polymerase domain (25–468, 444 aa in length) lost nearly 90% of prime editing efficiency, while another variant (24–474, 451 aa in length) retained 70% of the editing efficiency (Fig. 1e, Supplementary Fig. 8). To improve the activity of the truncated 451 aa RT, which is the shortest MMLV-RT variant to our knowledge, we first constructed 31 variants harboring mutations that were previously reported to enhance the MMLV-RT performance32. Using the minimally active 444 aa RT as a baseline, we identified eight mutations that individually increased the prime editing efficiency of the 444 aa RT (Supplementary Fig. 9). When implemented into the 451 aa RT variant, the D200C mutation increased the prime editing efficiency by 27% (Fig. 1f).
Finally, we focused on enhancing the electrostatic interactions between the MMLV-RT and the negatively-charged DNA/RNA hybrid guided by the crystal structure of XMRV-RT (PDB: 4HKQ)33, which shares high homology with our 451 aa MMLV-RT (Supplementary Fig. 10). We modified 44 residues within 10 Å of the DNA/RNA substrate individually to positively charged Arginine within the 451 aa MMLV-RT (D200C) (Supplementary Fig. 11) and discovered seven additional mutations that enhanced the editing efficiency. Through subsequent screening of these mutations in different combinations, we identified the double mutant 451 aa MMLV-RT (V101R + D200C), which demonstrated a 9% increase in BFP-to-GFP conversion efficiency compared to the single mutant 451 aa MMLV-RT (D200C) (Fig. 1g, Supplementary Fig. 12a–e). We also evaluated the performance of our engineered PEs at the human endogenous HEK3 locus, showing a consistent correlation of the editing efficiency with the reporter systems (Fig. 1h). Thus, our rationally engineered PE (EP3.61), featuring a truncated 451 aa MMLV-RT (V101R + D200C) and optimized NLS sequences, achieved similar endogenous editing efficiencies compared to PE2 with the full-length RT (Fig. 1h). Additionally, when coupled with the DAP eMPE array, EP3.61 can achieve 69% higher prime editing efficiency compared to PE2 (Fig. 1h). Collectively, we engineered a compact and efficient prime editing system with the shortest active MMLV-RT reported up to date, termed “prime editing with advanced kernel” (PEAK), that incorporates an N-terminal VirD2 NLS, a C-terminal SV40 NLS, a truncated 451 aa MMLV-RT with beneficial mutations V101R and D200C, and epegRNA in DAP eMPE array (Fig. 1h).
MPH-recruiting prime editors enable transcriptional activation on fluorescent reporters
To develop a gene activation system using PE, we incorporated the SAM system, a powerful RNA-guided programmable gene activator composed of a catalytically dead Cas9 (dCas9), activation sgRNA (agRNA) with two MS2-binding stem loops, and an MS2-p65-HSF1 (MPH) transcriptional activator18. Following MPH recruitment to the target loci through the MS2-binding stem loops, the SAM system attracts transcription factors and chromatin remodeling complexes for gene upregulation. We hypothesized that substituting the dCas9 of the SAM system with a nCas9 or a PE could still achieve sufficient gene activation (Fig. 2a). We first developed nine reporter variants with different half-lives of EGFP and copy numbers of protospacer targets. We used an agRNA with a 20-nt protospacer sequence to direct a nCas9 or PE, along with MPH, to the protospacer target region of the reporter for EGFP gene activation, which was subsequently quantified via flow cytometry.
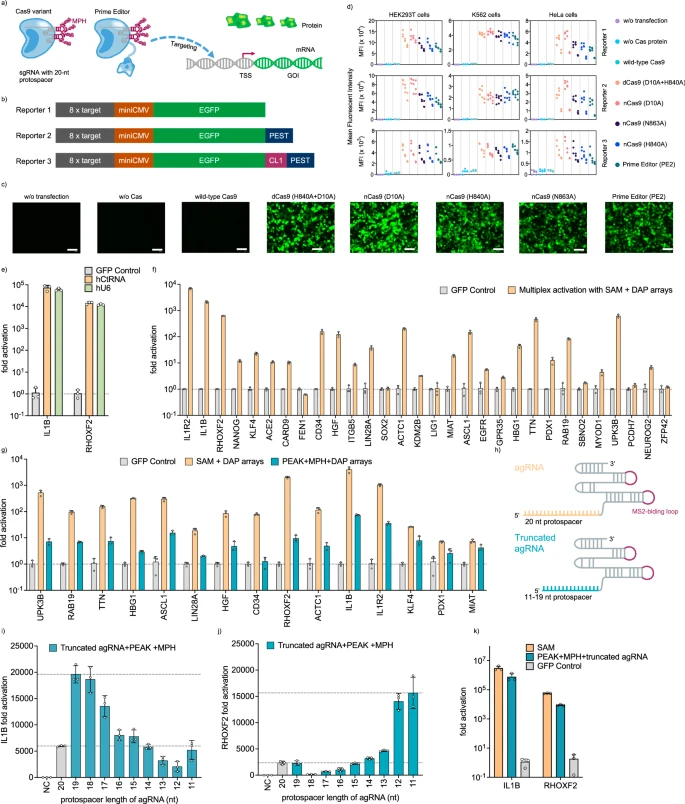
a Schematic of gene activation using either the Cas9 nickase variant or a prime editor. TSS transcription start site, GOI gene of interest. b Plasmid fluorescent reporters 1–3 with 8 × target protospacer, a miniCMV promoter, and green fluorescent proteins of varying half-lives. CL1 and PEST are degrons that can destabilize their fused proteins and half lives of EGFPs follows the hierarchy: EGFP-CL1-PEST < EGFP-PEST < EGFP. c Representative fluorescence microscope images showcasing the activation of Reporter 1 in HEK293T cells by Cas9 variants and prime editor from n = 3 biological replicates. Scale bar indicates 100 μm. d Flow cytometry analysis of gene activation across Reporters 1-3 in HEK293T, K562, and Hela cells, quantified by mean fluorescent intensity. e Activation of endogenous genes in HEK293T cells using dCas9 + MPH + agRNA, comparing agRNAs generated by either the hCtRNA promoter or the human U6 promoter. f Multiplex endogenous gene activation using dCas9, MPH, and DAP arrays encoding multiple agRNAs. g Comparison between multiplex gene activation by SAM + DAP and by PEAK + MPH + DAP. h Schematic of a full-length agRNA and a truncated spacer agRNA. i, j Activation of endogenous gene IL1B (i) and RHOXF2 (j) in HEK293T cells via PEAK, MPH, and truncated spacer agRNAs. k Endogenous IL1B and RHOXF2 gene activation using SAM system or PEAK with MPH and truncated agRNAs. A 19-nt-spacer and an 11-nt-spacer agRNAs were coupled with PEAK to activate endogenous IL1B and RHOXF2, respectively. Bars represent the mean ± s.d. from n = 3 independent biological replicates. Source data are provided as a Source Data file.
Reporters 1-3 were designed with eight copies of protospacer targets upstream of a miniCMV promoter that drives the expression of EGFP, EGFP-PEST, and EGFP-CL1-PEST, respectively (Fig. 2b). The protein degradation sequences PEST and CL1 were included to shorten the half-life of the fused proteins, thereby enhancing the fluorescent reporters’ signal-to-background performance34,35. In HEK293T cells, all evaluated nCas9 variants (D10A, H840A, and H863A) and PE2 successfully activated Reporters 1-3 when paired with the 20-nucleotide (nt) agRNA and MPH from the SAM system (Fig. 2c). Furthermore, when we extended our testing to different human cell lines, including K562 and Hela, we observed substantial activation of Reporters 1-3 with the nCas9 variants and PE2 compared to the non-transfected and no Cas plasmid transfection controls (Fig. 2d, Supplementary Fig. 13). Overall, PE2 retained an average of 77% GFP activation, while the nCas9 variants showed activities comparable to dCas9 (82–108%) (Fig. 2d, Supplementary Fig. 13). As expected, substituting dCas9 with wild-type Cas9 (wtCas9) resulted in only 6% GFP activation, likely due to double-stranded DNA cleavage (Fig. 2d, Supplementary Fig. 13).
To increase the stringency for gene activation, we designed Reporters 4-6 with only one protospacer target instead of eight (Supplementary Fig. 14a). Using these reporters, we observed that all nCas9 variants and PE2 still enabled significant gene activation, ranging from 72% to 102% EGFP fluorescence compared to dCas9 (Supplementary Fig. 14c, d). In contrast, wtCas9 retained only 19% of the activation capability. Finally, to assess the impact of nicking on either the sense or the antisense DNA strand on gene activation, Reporters 7-9 were designed with protospacer regions on the opposite strand compared to Reporters 4-6 (Supplementary Fig. 14b). We observed no significant difference in gene activation when nicking occurred on the sense or antisense DNA strand (Supplementary Figs. 14c, d, 10). Together, these results demonstrate that PE, a fusion of Cas9 nickase (H840A) and MMLV-RT, along with Cas9 nickases (D10A, H840A, and N863A), but not wtCas9, can effectively substitute dCas9 in the SAM system to activate gene expression in fluorescent reporter assays.
Efficient activation with MPH-recruiting prime editors on endogenous gene loci
Next, we aimed to determine if agRNAs expressed from the DAP array can effectively activate endogenous genes, which has not been previously reported. We expressed agRNAs targeting IL1B or RHOXF2 using either individual DAP arrays or a conventional U6 promoter, which were co-delivered with the dCas9-SAM system1. Compared to individual U6 promoters, we observed similar gene activation for both genes using the DAP array (with 75 bp hCtRNA as the promoter), while reducing the promoter length by 70% (Fig. 2e). Further expanding our approach, we assembled five DAP arrays each containing six agRNAs, allowing for 30-gene multiplexed activation using the dCas9-SAM system. Remarkably, 29 out of the 30 targeted genes were activated, with an average 404-fold increase in mRNA expression compared to the GFP transfected control as measured by RT-qPCR, demonstrating the DAP array’s capacity for efficient multiplex endogenous gene activation (Fig. 2f).
However, when dCas9 was substituted with PEAK in the SAM system, gene activation decreased by 89% on average across 15 tested endogenous genes (ranging from 99.5% to 43%) (Fig. 2g). We speculated that this reduced efficiency could be attributed to the DNA nicking activity of the nCas9 (H840A) in the PEAK system, as we also observed a 15% average reduction (up to 48%) in our EGFP reporter assay using nCas9 (H840A) or PE2 (Fig. 2d, Supplementary Figs. 13, 14, 15). Inspired by a truncated sgRNA design4,5,36 that effectively inactivates the nuclease activity of wtCas9 for efficient gene activation, we hypothesized that a similar truncation in the 20-nt protospacer of agRNA could also enhance endogenous gene activation efficiency by PEAK. Therefore, we constructed a series of truncated agRNAs (driven by a DAP array) with spacer lengths ranging from 8 to 20 nt (Fig. 2h). We then evaluated the gene activation capabilities of PEAK along with other Cas9 variants, including dCas9, wtCas9, nCas9(D10A), and nCas9(H840A), with truncated agRNAs (Fig. 2i, j, Supplementary Figs. 16, 17, 18). At the IL1B site in HEK293T cells, we found that a 19-nt agRNA demonstrated significantly improved activation, resulting in a 4,676-fold increase with PE2 and a 19,619-fold increase using PEAK. This represents a 280% and 328% improvement in activation capability compared to 20-nt agRNA paired with PE2 (1698-fold) or PEAK (5974-fold), respectively (Fig. 2i and Supplementary Fig. 17). Similarly, at the RHOXF2 site in HEK293T cells, an 11-nt agRNA enabled highly efficient gene activation (15,649-fold increase), while a 20-nt agRNA resulted in significantly lower activation (2,360-fold increase) with PEAK (Fig. 2j). Further, PEAK with truncated spacer agRNAs reached similar levels of activation compared to the dCas9-based SAM system with full-length spacer agRNAs on the IL1B and RHOXF2 genes (Fig. 2k). In HepG2 cells targeting the PDX1 gene, an agRNA with 11-nt protospacer allowed PEAK to achieve substantially higher gene activation (438-fold increase) compared to a 20-nt agRNA (83-fold increase), yielding results comparable to the dCas9 SAM system (534-fold increase) (Supplementary Fig. 18). Together, we demonstrate a viable strategy for efficient endogenous gene activation using PEAK and the DAP array, through the optimization of truncated agRNAs with spacer lengths ranging 11 and 19 nucleotides.
DAP shRNA array for efficient and scalable endogenous gene repression
To develop an efficient and orthogonal method for gene repression using PEAK and the DAP array, we aimed for a compact and modular approach that can independently silence multiple genes without affecting gene editing or activation. We initially adopted the CRISPR interference (CRISPRi) strategy37, utilizing the PE2 and a traditional sgRNA to inhibit the transcription of targeted genes (Fig. 3a). We designed an EGFP reporter and employed PE2 to repress the EGFP expression from the transfected plasmid in HEK293T cells (Fig. 3a, Supplementary Fig. 19). While PE2 was able to achieve up to 57% EGFP repression, it required laborious screening of the optimal protospacer (Fig. 3b). Only 17 of the 33 tested sgRNAs repressed the EGFP reporter expression by more than 50% (Fig. 3b). In addition, we observed no synergistic effects in EGFP repression when multiple sgRNAs were co-delivered (Supplementary Fig. 20). We also examined the more potent CRISPR repressor dCas9-KRAB-MeCP238, which outperformed PE2-mediated CRISPRi strategy, achieving an average of 88% EGFP repression with the top-performing sgRNAs (sgRNA6, 19, and 21) (Fig. 3d, Supplementary Figs. 20, 21). Although fusing PE with dCas9-KRAB-MeCP2 or other repressor proteins could potentially improve endogenous gene repression, the added repressors would not only interfere with gene activation, but also increase the protein fusion size, negating our goal of creating a compact and orthogonal gene perturbation system.
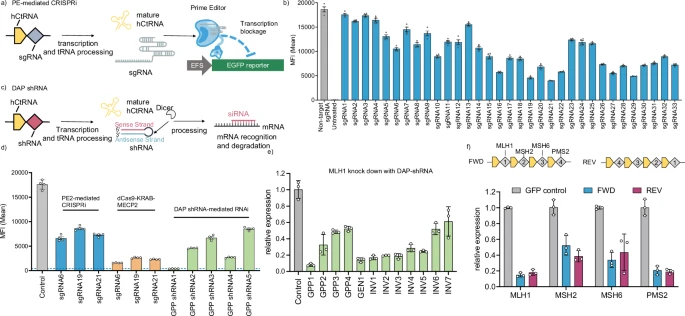
a Illustration of gene repression using PE2-mediated CRISPRi strategy targeting an EGFP reporter gene. b EGFP reporter repression with 33 different sgRNAs that span the EGFP gene. c Schematic of gene silencing achieved via shRNAs produced by the DAP array. d Comparative analysis of gene repression efficiencies using three different methods: PE2-mediated CRISPRi, dCas9-KRAB-MECP2 fusion protein, and DAP shRNA-mediated RNAi. GPP: Broad Institute GPP web portal. e Repression of the endogenous MLH1 gene using shRNAs designed by different web tools and expressed from DAP arrays. GEN GenScript siRNA design tool, INV InvivoGen siRNA Wizard. f Multiplex gene repression with DAP-shRNA array. FWD and REV are two DAP arrays encoding the same set of shRNAs in opposite order. Experiments were performed in HEK293T cells and analyzed using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) or flow cytometry. Bars represent mean ± s.d. from n = 4 (b, d) and n = 3 (e, f) independent biological replicates. Source data are provided as a Source Data file.
As an alternative strategy, RNAi is a well-established mechanism for gene silencing in human cells19,39. shRNAs, typically 48 base pairs in length, have been known to achieve highly efficient gene repression40,41,42 and present a viable option for incorporation into our DAP array. Given our previous success in using the DAP array to express short RNAs for gene editing and activation, we hypothesized that shRNAs could also be integrated into the DAP array for orthogonal gene repression (Fig. 3c). To test this hypothesis, we selected the top five shRNAs (GPP shRNA1-5) generated by the genetic perturbation platform (GPP) webtool to target the EGFP transcript. GPP shRNA1 repressed EGFP expression by more than 98%, surpassing the efficiency of protein-based transcription repression systems (Fig. 3d). Additionally, as the 3’ end overhang may affect shRNA’s gene silencing function43, we tested GPP shRNA1-expressing DAP arrays with various poly-A tail sequence lengths and found no influence on gene repression efficiency (Supplementary Fig. 22).
Next, we designed a dozen shRNAs targeting the endogenous MLH1 gene, which encodes a key protein of cellular DNA repair mechanisms44, to validate the efficacy of shRNAs generated by the DAP array in repressing the endogenous gene in human cells. The GPP1 shRNA achieved the most efficient endogenous MLH1 gene knockdown (92%), outperforming those designed by GenScript (GEN) or InvivoGen (INV) (Fig. 3e). To further examine the multiplexed gene repression using our system, we designed a DAP array expressing four shRNAs designed by GPP, each targeting a key gene involved in the DNA mismatch repair (MMR) pathway – MLH1, MSH2, MSH6, and PMS2, potentially mediating prime editing in MMR-proficient cells44. The multiplexed DAP shRNA array demonstrated highly efficient gene repression, with knockdown efficiencies of MLH1, MSH2, MSH6, and PMS2 at 85%, 48%, 66%, and 79%, respectively (Fig. 3f). In addition, the order of shRNAs in the DAP array did not affect the knockdown efficiency (Fig. 3f). When co-delivered with PEAK and an eMPE array targeting the endogenous human HEK3 locus, the multiplex DAP shRNA array increased the editing efficiency by 1.2-fold in partially MMR-deficient HEK293T cells (Supplementary Fig. 23). This result is consistent with previous reports for the HEK3 locus in HEK293T cells when co-delivering MLH1dn, and the effect of MMR repression on PE efficiency through shRNA arrays may be further enhanced in MMR-proficient cell lines44. Together, these findings underscore the DAP-derived shRNA array as a minimal, versatile RNA module for efficient gene repression, functioning independently of gene editing and activation by PEAK.
Simultaneous genetic perturbations using the DAP array, PEAK, and MPH
Thus far, we have established a minimal and versatile genetic perturbation technology (mvGPT) comprising a diverse RNA-encoding DAP array, PEAK, and MPH to orchestrate endogenous gene editing, activation, and repression. To explore its potential for simultaneous gene perturbations, we targeted three distinct genetic loci relevant to Wilson’s disease, Type I diabetes, or transthyretin amyloidosis (Fig. 4a). Wilson’s disease, commonly caused by a c.3207C>A; p.H1069Q mutation in the ATP7B gene45, leads to excessive copper accumulation in the body and potential liver failure46. For Type I diabetes, activating the PDX1 gene can induce hepatocyte transdifferentiation into pancreatic beta-like insulin-producing cells, increasing insulin levels and lowering glucose levels in the blood47,48. Finally, hereditary transthyretin amyloidosis is a severe disease caused by mutations in the transthyretin (TTR) gene, resulting in extracellular amyloid deposition and multiple organ dysfunctions49,50. Reducing transthyretin levels has been shown to be an effective therapeutic approach to manage this disease39.
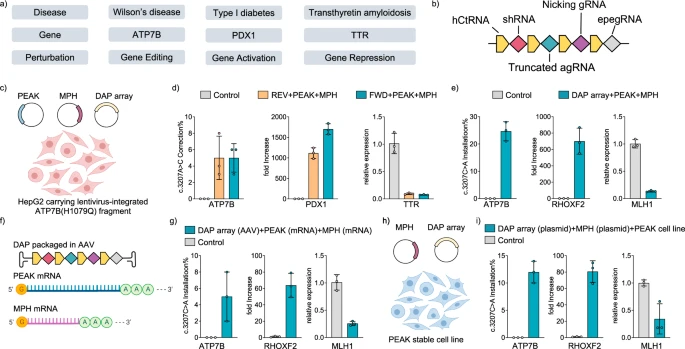
a Schematic of a hypothetical complex genetic disease model involving Wilson’s disease, Type I diabetes, and Transthyretin amyloidosis. Treatment of the disease model requires orthogonal editing of the ATP7B gene, activation of the PDX1 gene, and repression of the TTR gene. b Design of a DAP array encoding a shRNA for gene silencing, a truncated agRNA for gene activation, and a ngRNA and a epegRNA for gene editing. c, d Therapeutic genetic perturbation in HepG2 disease cell line transfected by plasmids encoding the DAP array, PEAK, and MPH. REV: the direction of DAP array was reversed as compared to FWD DAP array. e Genetic perturbation in HEK293T cells transfected with plasmids encoding the DAP array, PEAK, and MPH to install the c.3207C>A mutation in the ATP7B gene, upregulate the expression of RHOXF2 gene, and silence the MLH1 gene. f, g Combinatorial delivery of the DAP array (AAV), PEAK (mRNA), and MPH (mRNA) into HEK293T cells. h, i Combinatorial delivery using plasmids for the DAP array and MPH, and lentivirus for PEAK. Controls were untreated cells. A stable cell line expressing PEAK was established before introducing the DAP array and MPH via plasmid transfection. Gene editing outcomes were analyzed by Sanger sequencing and transcriptional regulations were analyzed by RT-qPCR. Error bars represent mean ± s.d. from n = 3 independent biological replicates. Source data are provided as a Source Data file. c, f, h Created in BioRender. Yuan, Q. (2023) BioRender.com/b09r397.
To simultaneously alleviate conditions associated with these genetic diseases, we created a DAP array encoding an eMPE array (to correct the ATP7B H1069Q mutation via a c.3207A>C substitution), an optimized 11-nt spacer agRNA (to activate the PDX1 gene), and the shRNA from the medication Patisiran49 (to repress the TTR gene), which outperformed all other shRNAs that we designed and tested (Fig. 4b, Supplementary Figs. 18, 24). As liver cells are closely involved in the cause and treatment of all three diseases, we used the human hepatoma cell line HepG2 as our testing platform. We first established a HepG2 stable cell line with a short sequence containing the pathogenic ATP7B c.3207C>A; p.H1069Q mutation via lentiviral transduction. Subsequently, we transfected these HepG2 cells with plasmids encoding the designed DAP array, PEAK, and MPH (Fig. 4c). The results demonstrated a 5% ATP7B c.3207A>C correction, up to a 1700-fold activation of the PDX1 gene, and a 93% repression of the TTR gene in the HepG2 stable cell line (Fig. 4d). To further validate the capabilities of mvGPT for multiplexed gene perturbations, we constructed another DAP array with different targets. This array encoded for an eMPE array to install the Wilson’s disease-causing ATP7B c.3207C>A; p.H1069Q mutation in situ, a truncated agRNA with an 11-nt spacer to activate the RHOXF2 gene, and an shRNA to silence the MLH1 gene. When tested using plasmid delivery, we observed a 25% prime editing efficiency for ATP7B c.3207C>A installation, a 700-fold gene activation of RHOXF2, and an 87% repression of MLH1 gene expression in HEK293T cells (Fig. 4e, Supplementary Fig. 25). Using single cell analysis, we observed simultaneous prime editing, gene activation, and repression in 11 out of 16 treated single cells (Supplementary Fig. 26). In addition, when PEAK and the DAP array were delivered without MPH, we observed prime editing and gene repression without activation. Delivering MPH and the DAP array without PEAK resulted in gene repression only (Supplementary Fig. 27). These results suggest that mvGPT perturbations are orthogonal and do not exhibit cross-reactivity.
Finally, we explored alternative delivery modalities beyond plasmids for mvGPT. We first prepared the DAP array, PEAK, and MPH as messenger RNAs (mRNAs). Interestingly, delivering the DAP array as mRNA did not produce functional RNAs in cells, likely due to its inability to enter the nucleus for pre-tRNA processing51. As a result, instead of delivering all three components as mRNAs, we decided to package the DAP array into an AAV vector (Fig. 4f). Four days after transducing HEK293T cells with AAV carrying the DAP array, we transfected cells with MPH and PEAK mRNAs. This strategy led to a 5% prime editing efficiency for installing the ATP7B c.3207C>A; p.H1069Q mutation, a 64-fold gene activation of RHOXF2, and a 75% gene repression of MLH1 (Fig. 4g). Furthermore, we packaged PEAK into a single lentiviral vector with a puromycin resistance gene and transduced HEK293T cells for selection. After establishing a stable cell line expressing PEAK, we transfected cells with plasmids encoding the DAP array and MPH (Fig. 4h). This approach achieved a 12% prime editing efficiency for installing the ATP7B c.3207C>A; p.H1069Q mutation, an 81-fold upregulation of RHOXF2, and a 66% gene repression of MLH1 (Fig. 4i). These results demonstrated the effective combinatorial viral and non-virial delivery of the DAP array, PEAK, and MPH for simultaneous endogenous genetic perturbations, including gene editing, gene activation, and gene repression.
Discussion
Here, we present mvGPT, a streamlined and modular platform to perform simultaneous and orthogonal endogenous gene editing, activation, and repression in human cells. The mvGPT comprises three primary molecular modules: the DAP array, PEAK, and MPH. Functioning as the central component of mvGPT, the DAP array utilizes an engineered hCtRNA as a robust RNA Pol III promoter, facilitating the tandem expression of programmable small RNAs. These RNAs, released by cellular tRNA processing mechanisms, enable orthogonal genetic perturbations. The absence of lengthy promoters in the DAP array reduces the delivery payload, enabling high scalability and multiplexity with mvGPT.
Gene editing in the mvGPT platform is accomplished when PEAK complexes with pegRNA and ngRNA, both products of the DAP array. PEAK, the prime editing system with advanced kernel, includes a truncated 451 aa MMLV-RT, the shortest MMLV-RT variant up to date derived from PE2, with enhancing mutations D200C and V101R, an engineered N-terminal VirD2 NLS, and a C-terminal SV40 NLS. PEAK can effectively pair with epegRNA encoded in the DAP array to enable enhanced prime editing activity. In contrast to prior Cas nuclease-based technologies, mvGPT employs prime editing to modify the context of the genome without causing DSBs, thus avoiding error-prone and cytotoxic DNA repair mechanisms and enhancing the safety of potential therapeutic applications. The gene activation function operates orthogonally to gene editing by leveraging the MPH activator, which is recruited to PEAK with a site-dependent and spacer-tailored (11-19 nt) agRNA. Finally, gene repression is independently achieved by shRNA produced by the DAP array. Importantly, gene repression mediated by the DAP array-generated shRNA avoids the incorporation of additional proteins, thereby minimizing interference with the gene editing and activation process. We demonstrate that mvGPT is compatible with both viral and non-viral delivery modalities for enhanced applications. The compact size of mvGPT positions it as a useful tool for studying complex genomic functions or complex genetic diseases where precision and tunable perturbations on both the genome and transcriptome are required.
In summary, mvGPT represents a compact and versatile molecular technology that enables effective simultaneous and orthogonal gene editing, activation, and repression in human cells, providing better support for the genetic interrogation of complex biology, the study of complex genetic diseases, and the development of human gene therapy. Future improvement could include incorporating recently engineered PEs with various RT domains, mutations, and effector fusions that enhance editing efficiencies44,52,53. To minimize the risk of off-target activation by spacer-truncated agRNAs, off-target analysis is essential to ensure that agRNAs do not bind to unintended sites near downstream transcription start sites (TSS)18. We envision that the mvGPT could leverage recently reported proximal dead single guide RNAs and truncated sgRNAs to modify chromatin structure and activate edited genes54,55. This could be combined with shRNA-mediated knockdown of endogenous DNA repair mechanisms44 to further improve PE efficiency. Additionally, the role of the RT domain in endogenous gene activation achieved by PEAK, when used with truncated agRNAs, might require further investigation. Finally, the discovery and adaptation of potent mammalian endogenous aptamer-activator systems to the mvGPT platform could further reduce its size. Applications of mvGPT may extend to primary cells and animal models of complex genetic diseases, underscoring its therapeutic promise.
Methods
Molecular cloning
Plasmids, sgRNAs, and primers were designed and generated using Benchling. DNA templates for polymerase chain reaction (PCR) were from previously established plasmids, Addgene plasmids, or synthesized fragments (IDT, gBlock). Standard PCR amplification was performed using 2 × Phanta Max Master Mix (Vazyme, P525) for DNA fragment or vector amplification. The resulting fragments were purified by gel extraction and assembled through Gibson Assembly Master Mix (New England Biolabs, E2611L) or Golden Gate Assembly with BsaI-HFv2 (New England Biolabs, R3733S) or Esp3I (Thermo Fisher Scientific, ER0451) and T4 DNA Ligase (New England Biolabs, M0202S). DNA assembly products were transformed into 10 µl Stbl3 competent cells generated by Mix and Go! E. coli Transformation Kit and Buffer Set (Zymo research, T3001) and plated on agar plates supplemented with 100 µg/ml ampicillin. Plasmids were obtained via DNA miniprep using the QIAquick PCR Purification Kit (Qiagen) and DNA spin column (Epoch Life Science). Typical PCR reactions (20 µl) included 1 µl template (1–10 ng/µl), 2 µl 10 µM primer pair, 7 µl ultrapure (Millipore) or distilled water, and 10 µl 2 × Phanta Max Master Mix (Vazyme, P525). Annealing temperatures, typically set at 60 °C, could be adjusted between 55 °C and 65 °C for optimal amplification yield. Long DNA fragments (>10 kb) could be amplified with high-fidelity via 25-cycle PCR. Plasmid sequence information and examples for constructing DAP arrays can be found in the source data table.
Cell culture
All cells were maintained and passaged in 10 mL TC treated cell culture dishes with vents (Greiner Bio-One, 639160). HepG2 cells (ATCC, HB-8065) were cultured in Eagle’s Minimum Essential Medium (EMEM) (ATCC, 30-2003) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco, 10437028) and 1% (v/v) penicillin-streptomycin (Pen-Strep) (Gibco, 15140122). HEK293T cells (ATCC, CRL-3216) and HeLa cells (ATCC, CCL-2) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) plus GlutaMAX (Gibco, 10569044) supplemented with 10% (v/v) FBS (Gibco, 10437028) and 1% (v/v) Pen-Strep (Gibco, 15140122). K562 (ATCC, CCL-243) was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium plus GlutaMAX (Gibco, 61870036) supplemented with 10% (v/v) FBS (Gibco, 10437028) and 1% (v/v) Pen-Strep (Gibco, 15140122). Cells were incubated at 37 °C with 5% CO2 and passaged upon reaching 80-90% confluency. Cells were authenticated by the supplier using STR (short tandem repeat) analysis.
Transfection
Cells with low passage number (1–10, freshly thawed counted as 0) were passaged every other day and counted using a Countess II FL Automated Cell Counter (Thermo Fisher Scientific) before seeding for transfection. The seeded plate was pre-incubated at room temperature on a flat surface for 15 min before being placed into the incubator to reduce the edge effect and avoid unevenly seeded cells. For fluorescent reporter relevant assays, cells were plated at 2 × 104 cells (Reporters 1-9, BFP v2 reporter cell line, EFS-EGFP and BFP reporters) or 0.75 × 104 cells (3-color reporter cell line) per 100 µl culture medium per well in 96-well plates (Corning, 3598) 16–18 h before transfections. For gene editing, gene activation, and gene repression assays, cells were plated at 0.75–2 × 104 (HEK293T cells) or 1.5 × 104 (HepG2 cells) per 100 µl culture medium per well in poly-D-lysine coated plates (Corning, 356690) 16 h before transfections. Transfection reagents including PEI Max (1 mg/ml, PH = 7.1, Polysciences), Lipofectamine 2000 (Invitrogen, 11668019), Lipofectamine 3000 (Invitrogen, L3000001), Lipofectamine MessengerMax (Invitrogen LMRNA001) were used. In fluorescent reporter assays, both PEI Max and Lipofectamine 2000 were used. In gene editing, gene activation, and gene repression assays, both Lipofectamine 2000 (for HEK293T cells) and Lipofectamine 3000 (for HepG2 cells) were used. PEI Max transfection for each well of a 96-well plate was optimized, briefly, 100–250 ng DNA and 0.5 µl PEI Max were diluted in 5 µl OptiMEM I Reduced-Serum Medium (Gibco, 31985062), respectively. Then, 5 µl diluted DNA was mixed with 5 µl diluted PEI Max for 5 min before being added into each well (Supplementary Fig. 28). Lipofectamine 2000 transfections were performed following the same reagent protocol, but using 0.5 µl Lipofectamine 2000. Lipofectamine 3000 transfections were performed following the same reagent protocol, but using 0.5 µl Lipofectamine 3000, 0.5 µl P3000, and up to 450 ng DNA for each well of a 96-well plate. Specifically, for reporter gene activation assay, 150 ng plasmid of sgRNA with MS2 stem loops, 150 ng plasmid of Cas9 variant or prime editor, 150 ng plasmid of MPH, and 50 ng fluorescent reporter plasmid were transfected in HEK293T, K562 and Hela cells using Lipofectamine 2000. For prime editing assay to be quantified by sequencing, 225 ng plasmid of prime editor variant and 75 ng plasmid of MPE DAP array were transfected in HEK293T cells using Lipofectamine 2000. For prime editing assay to be quantified by flow cytometry, 150 ng plasmid of prime editor and 50 ng plasmid of fluorescent reporter were transfected in HEK293T cells using PEI Max. For gene repression assay, 50–500 ng plasmid of DAP-shRNA was transfected using Lipofectamine 2000 (HEK293T cells) or 3000 (HepG2 cells). For genetic perturbation assay, 150 ng plasmid of PEAK, 150 ng plasmid of MPH, and 150 ng plasmid of the DAP array were transfected using Lipofectamine 2000 (HEK293T cells) or 3000 (HepG2 cells). For multiplex gene activation assay in HEK293T cells, 150 ng plasmid of PEAK/dCas9/PE2, 150 ng plasmid of MPH, and 200 ng pooled plasmids of DAP array were transfected. A plasmid expressing EGFP was used as control in quantitative reverse transcription PCR (RT-qPCR) assays. Dose-relevant assays comparing MPE and eMPE were performed by only changing the amount of DAP array, with no filler plasmid being used. For AAV and mRNA combinatorial viral and non-viral delivery, 4 days after the AAV transduction of the DAP array, 1500 transduced HEK293T cells were transfected with 150 ng MPH and 200 ng PE mRNA using Lipofectamine MessengerMax, following the manufacturer’s protocol. Cellular DNA and RNA were extracted 3 days after transfection for gene editing, gene activation, and gene repression analysis.
Genomic DNA extraction
The culture medium was carefully aspirated from each well for HEK293T, Hela, and HepG2 cells. Next, 100 µl of freshly prepared lysis buffer [10 mM Tris-HCl, pH7.5, 0.05% SDS, 25 µl/ml proteinase K (Thermo Fisher Scientific)] was added to each well of the 96-well plate. The samples were incubated at 37 °C for 5 min and then heat-inactivated at 80 °C for 30 min. Genomic DNA lysate was used immediately or stored at 4 °C.
Flow cytometry
Approximately 48–60 h after transfection, the fluorescence of each well was imaged using the EVOS FLoid Imaging System (Thermo Fisher Scientific). For flow cytometry sample preparation, the culture medium of each well was gently aspirated, followed by the addition of 100 µl TrypLE Express (Thermo Fisher Scientific, 12605028) per well of a 96-well plate. The samples were then incubated at 37 °C for 5 min before dilution with 150 µl/well culture medium. High-throughput flow cytometry was performed using the Sony SA3800 Flow Cytometer, and the data was analyzed using FlowJo 10.8.1 (FlowJo, LLC). Cells were gated by forward versus side scatter (FSC vs. SSC) plot to identify cell population and exclude debris, forward scatter height versus forward scatter area (FSC-H vs. FSC-A) plot for doublet exclusion, and FSC-H or histogram vs. EGFP-A, BFP-A, or mCherry-A plot to reflect fluorescence signals. All represented samples had at least three biological replicates. Data are representative of at least 5000 gated events per condition.
Fluorescence-activated cell sorting
The Sony MA900 Cell sorter was used for performing fluorescence-activated cell sorting (FACS) experiments. Cells were gated by forward versus back scatter (FSC vs. BSC) plot to identify cell population and exclude debris. Forward scatter height versus forward scatter area (FSC-H vs. FSC-A) plot was used for doublet exclusion, and FSC-H or histogram vs. BFP-A plot to reflect fluorescence signals. In developing HEK293T, K562, and Hela stable cell lines integrated with BFP or BFP v2 (BFP gene and the DAP array to enable H66Y prime editing), the cell population with the top 5% BFP fluorescent signal was sorted from cells transduced with 100 × MOI (Supplementary Fig. 29).
Targeted amplicon sequencing and data analysis
Genomic regions surrounding each target locus were amplified, purified, quantified, and sent for Sanger sequencing (Epoch Life Science) or next-generation sequencing (NGS) (Amplicon-EZ, Genewiz). Partial Illumina adapters provided by Amplicon-EZ were added to the 5’ end of each forward and reverse primer. A typical 10 µl PCR reaction was conducted using 0.5 µmol of each forward and reverse primer (IDT), 1 µl genomic DNA extract, and 5 µl 2 × Phanta Master Mix (Vazyme), with a 60 °C annealing temperature and 35-cycle amplification. All primer pairs successfully amplified the desired fragments, verified by DNA electrophoresis in a 1% agarose gel. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and DNA spin column (Epoch Life Science). For Sanger sequencing, each amplicon was eluted in 20 µl ultrapure water (Milipore) and quantified by NanoDrop OneC (Thermo Scientific). The sequencing premix (15 µl) was prepared by adding 1 µl diluted DNA (10–20 ng) and 2.5 µl 10 µM sequencing primer to 11.5 µl ultrapure water. For NGS, multiple amplicons were pooled, purified, and eluted in 30 µl of ultrapure water, then quantified first by NanoDrop OneC (Thermo Fisher Scientific) to adjust the DNA concentration to 60–80 ng/µl, and subsequently by the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) to obtain ~500 ng amplicon in 25 µl ultrapure water for Amplicon-EZ. Sanger sequencing results were analyzed using EditR (http://baseeditr.com/). NGS results were analyzed using CRISPResso2 (http://crispresso2.pinellolab.org). Sanger sequencing and NGS data were visualized using the GraphPad Prims 9.4.1.
RNA extraction
RNA was extracted using the Quick-RNA Miniprep (Zymo Research, R1055) according to the manufacturer’s instructions. Three days post-transfection, the culture medium was removed, and 300 µl of RNA Lysis Buffer was added to each well in a 96-well plate. The cell lysate was cleared by centrifugation at 15,800 × g for 1 min, after which the supernatant was transferred to a Spin-Away Filter in a collection tube and centrifuged at 15,800 × g for 1 min to remove most genomic DNA. The flow-through was collected in a separate 1.5 mL microcentrifuge tube, and an equal volume of 100% ethanol was added and mixed thoroughly. This mixture was then transferred to a Zymo-Spin IIICG column in a collection tube and centrifuged at 15,800 × g for 30 s. After discarding the flow-through, 400 µl RNA Prep Buffer was added to the column, and the sample was centrifuged at 15,800 × g for 30 s. The flow-through was discarded again, followed by the addition of 700 µl RNA Wash Buffer to the column and centrifugation at 15,800 × g for 30 s. Another wash step was performed using 400 µl RNA Wash Buffer, followed by centrifugation at 15,800 × g for 2 min to ensure complete removal of the wash buffer. For RNA elution, 50 µl DNase/RNase-free water was added to the column and centrifuged at 15,800 × g for 1 min. The eluted RNA was either used immediately or stored at −80 °C.
Reverse transcription (RT)
RT was carried out using HiScript III All-in-one RT SuperMix Perfect for qPCR (Vazyme, R333-01) following the manufacturer’s instructions. A 20 µl reaction mix was prepared, which included 4 µl of 5 × All-in-one qRT SuperMix, 1 µl Enzyme Mix, and 15 µl of the RNA elute with the lowest concentration measured by NanoDrop OneC (Thermo Scientific). An equivalent amount by mass of RNA was added for samples in the same batch with higher concentration, and RNase-free ddH2O was used to make up the 20 µl volume. The reaction mix was incubated at 50 °C for 15 min, followed by 85 °C for 5 s. The resulting coding DNA (cDNA) was either used immediately for qPCR or stored at −20 °C.
Endogenous gene activation sgRNA design
The NCBI reference sequence database (RefSeq) accession number of a transcript of the gene of interest (GOI) was obtained from UCSC Genome Browser using its table browser. This RefSeq accession number was then provided to Benchling to import the sequence of the GOI along with annotations of functional elements. sgRNAs with MS2 stem loops were designed to target the 300-bp region upstream of the TSS of GOI.
Quantitative polymerase chain reaction (qPCR)
qPCR primers were selected from the “qPCR primers” track in the UCSC genome browser56. qPCR was performed using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Q712-02) following the manufacturer’s protocol. In brief, a 20 µl reaction mix was prepared to contain 10 µl of 2 × qPCR master mix, 7 µl of ddH2O, 0.5 µl of 10 µM Forward Primer, 0.5 µl of 10 µM Reverse Primer, and 2 µl cDNA template. The qPCR was conducted on a Bio-Rad C1000 Touch Thermal Cycler with a CFX96 Real-Time System or a Applied Biosystems QS 12 K Flex using the following program: Stage 1-Initial Denaturation, Rep 1, 95 °C, 2 min; Stage 2-Cycling Reaction, Rep 45, 95 °C for 5 s followed by 60 °C for 30 s; Stage 3-Melting Curve, Rep 1, 95 °C for 5 s, followed by 65 °C to 95 °C ramp at 0.5 °C/cycle. Data were extracted using Bio-Rad CFX Maestro 1.1 (Version 4.1.2433.1219) or using the QuantStudio 12 K Flex Software v1.6. Data analysis followed the Delta-Delta Ct method.
Single-cell analysis
HEK293T cells were seeded in a 12-well plate and transfected with PEAK, the DAP array with MPH, and pEGFP-N1 at the ratio of 4:4:1, or pEGFP-N1 only as control. One day before the single cell picking, cells were re-seeded on the poly-lysine coverslip (Corning 354086). The coverslip was removed and submerged in artificial cerebrospinal fluid (aCSF, pH 7.3) containing 126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 11.1 mM glucose, and 21.4 mM NaHCO3) saturated with 95% O2 and 5% CO2. Coverslip was transferred to a recording chamber and the cells was identified and excised under a fluorescence-equipped dissecting microscope. Single EGFP-labeled cells were visualized using epifluorescence and IR-DIC imaging on an upright microscope equipped with a moveable stage (MP-285, Sutter Instrument). Single cells were manually picked up by the pipette for RNA extraction, reverse transcription, pre-AMP, and qPCR using the Ambion Single-Cell-to-CT qRT-PCR Kit (Ambion, Life Technologies) according to the manufacturer’s instruction. Genomic editing efficiencies were analyzed by sequencing cDNA.
mRNA in vitro transcription
PEAK and MPH mRNAs were transcribed in vitro using the HiScribe T7 ARCA mRNA Kit (with tailing) (New England Biolabs, E2060S) with modified nucleotides following the manufacturer’s instructions. A DNA template containing a T7 promoter upstream of the GOI was prepared via standard PCR using 2 × Phanta Max Master Mix (Vazyme, P525) and purified through gel electrophoresis. A 20 μl IVT reaction was set up with 1 μg of DNA template, 10 µl of ARCA/NTP Mix (2X), 2.5 µl of 5mCTP(10 mM), 2.5 µl of Pseudo-UTP (10 mM), 2 μl of T7 RNA Polymerase Mix, and ddH2O filled to 20 µl. The reaction was mixed gently and incubated at 37 °C for 30 min. Then, 2 µl of DNase I was added, mixed well, and incubated at 37 °C for 15 min. Afterward, 5 μl of Poly(A) Polymerase Reaction Buffer (10X), 5 µl of Poly(A) Polymerase, and 20 µl ddH2O were added directly to the 20 µl IVT reaction, mixed gently, and incubated at 37 °C for 30 min.
mRNA purification
The transcribed mRNA was purified by LiCl precipitation using materials provided in the HiScribe T7 ARCA mRNA Kit (with tailing) (New England Biolabs, E2065S) following the manufacturer’s instructions. Briefly, 25 µl LiCl solution was added to the 50 µl tailing reaction mix, mixed well, and incubated at −20 °C for 30 min. Then, the mixture was centrifuged at 4 °C for 15 min at maximum speed to pellet the RNA. The supernatant was carefully removed, and the RNA pellet was rinsed with 500 μl of cold 70% ethanol, followed by centrifugation at 4 °C for 10 min. The ethanol was carefully removed, and any residual liquid was eliminated using a sharp tip. The RNA pellet was air-dried and resuspended in 50 μl of 0.1 mM EDTA RNA storage solution. The RNA was heated at 65 °C for 5–10 min to ensure complete dissolution and mixed well. mRNA quality and concentration were assessed using Nanodrop OneC (Thermo Scientific). Purified mRNA was either immediately used or was aliquoted and stored at −20 °C or below until further use.
Lentivirus and AAV production
Low passage HEK293T cells were seeded at 5 × 106 cells per 10 ml culture media [10% v/v FBS (Gibco, 10437028), 90% v/v DMEM plus GlutaMAX (Gibco, 10569044), and penicillin-streptomycin (Gibco, 15140122) diluted to 100 units/mL and 100 µg/mL, respectively] per 10-cm cell culture dish (Greiner Bio-One, 639160) 16 h before transfection. For lentivirus production per 10-cm dish, 5 μg of transfer vector plasmid containing the construct of interest, 2.5 μg of pMD2.G envelope plasmid (Addgene, #12259), and 4.5 μg of psPAX2 packaging plasmid (Addgene, #12260) were added into 260 μl of serum-free DMEM in a 50-ml tube, followed by addition of 78 μl PEI Max (1 mg/ml, PH = 7.1, Polysciences), vortexed, and then incubated at room temperature for 10 min. The transfection mixture was then diluted with 10 ml of culture medium to replace the old medium from the 10-cm dish. After 48 h, the full volume of supernatant was used directly or collected in a 15-ml tube and centrifuged at 3200 × g for 5 min at room temperature to remove the cell debris, clarified through a 0.45 μm PVDF filter (Millipore) and concentrated using PEG virus precipitation kit (Biovision) with an optimized protocol. Briefly, 2.5 ml of PEG solution was added to the 10 ml supernatant, inverted evenly, and refrigerated at 4 °C for 24 h. The mixture was then centrifuged at 3200 × g and 4 °C for 30 min, followed by several rounds of aspiration and centrifugation to entirely remove the supernatant from the precipitated white pellet. Lastly, the pellet was suspended in 80 μl of virus resuspension solution. The process for AAV production was similar to that of lentivirus production except for the plasmid used. For each 10-cm dish, 3 μg of vector plasmid, 5 μg of pHelper plasmid (Cell Biolabs), and 4 μg of AAV1-Rep-Cap plasmid (Addgene, #112862) were transfected. The freshly prepared lentivirus or AAV were immediately used for transductions.
Transduction
Low passage cells were seeded at 1500–2 × 104 cells per 100 μl culture medium per well in a 96-well poly-D-lysine coated plate (Corning, 356690) and pre-incubated at room temperature for 15 min, followed by the addition of freshly prepared lentivirus or AAV, and then placed into the incubator. Cells were transduced at different multiplicity of infection (MOI). When developing HEK293T, K562, and Hela cells reporter stable cell lines expressing BFP or BFP v2 (BFP with the DAP array to enable H66Y prime editing), transducing 2 × 104 cells/well with 0.1, 1, and 10 µl/well lentivirus concentrate represent 1 × MOI, 10 × MOI, and 100 × MOI, respectively. To develop HEK293T stable cell line expressing 3-color reporters, 100 µl lentivirus concentrate was added to 2 × 104 cells/well. When developing HEK293T stable cell line expressing PEAK, 2 × 104 cells/well were transduced with 20–40 µl lentivirus concentrate. 24 h after lentiviral transduction, 1 μg/ml puromycin (Thermo Fisher Scientific, J67236.8EQ) were supplemented into cell culture media to initiate puromycin selection. Once cells in the 96-well reached >90% confluency, they were dissociated and replated into a 10-cm cell culture dish with 10 mL culture media containing 1 μg/ml puromycin. Transduced cells were used in downstream experiments such as FACS or genomic editing once they reached >80% confluency. For AAV transduction, HEK293T cells were seeded at 1500 cells per 100 μl culture medium per well in the 96-well poly-D-lysine coated plate (Corning, 356690), pre-incubated at room temperature for 15 min, transduced with 30 µl AAV (encoding the DAP array) concentrate, and placed into the incubator. Three days after AAV transduction, cells in each transduced well were transfected with the PEAK and MPH mRNAs.
Prime editing gRNA design
PegRNAs and nicking gRNAs were designed using PrimerDesign57 (https://drugthatgene.pinellolab.partners.org/). Non-interfering nucleotide linkers between a pegRNA and the 3’ motif was designed using pegLIT26 (https://peglit.liugroup.us).
Endogenous gene repression shRNA design
shRNAs in the DAP array were designed using the Broad Institute GPP web portal (https://portals.broadinstitute.org/gpp/public/) or GenScript siRNA Target Finder (https://www.genscript.com/tools/sirna-target-finder) or InvivoGen siRNA Wizard Online Tool58 (https://www.invivogen.com/sirna-wizard).
Statistics and reproducibility
Values were reported as mean ± SD. Groups were compared using the unpaired two-tailed t-test or the nested one-way ANOVA with Dunnett’s multiple comparisons test. The solid lines and dashed lines of the violin plot represent the quartiles and median. Biologically independent experiments reported here were performed by different researchers using separate splits of the mammalian cell type used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genomic sequencing raw reads are available at the NCBI SRA database under accession code PRJNA955386. Plasmids for DAP array assembly are available at https://www.addgene.org/Xue_Gao/. Source data are provided with this paper.
Code availability
HTS reads were analyzed using crispresso2 (http://crispresso2.pinellolab.org). In the rational engineering, Pymol (Ver 2.5)59 was used with “select S1 within X of S2” which gives atoms in S1 that are within X Angstroms of any atoms in S2, to determine residues within 5/7/10 Å of the DNA-RNA duplex in the crystal structure of 451 aa XMRV-RT (PDB: 4HKQ)33.
Change history
11 February 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-56839-1
References
Yuan, Q. & Gao, X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat. Commun. 13, 2771 (2022).
McCarty, N. S., Graham, A. E., Studená, L. & Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 11, 1281 (2020).
Gao, Y. et al. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 13, 1043–1049 (2016).
Dahlman, J. E. et al. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 33, 1159–1161 (2015).
Kiani, S. et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 12, 1051–1054 (2015).
Campa, C. C., Weisbach, N. R., Santinha, A. J., Incarnato, D. & Platt, R. J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 16, 887–893 (2019).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B. & Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930 (2018).
Ihry, R. J. et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946 (2018).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Daniel, T. C., Zeng, H., Osikpa, E. C. & Gao, X. Revolutionizing genetic disease treatment: Recent technological advances in base editing. Curr. Opin. Biomed. Eng. 28, 100472 (2023).
Farzadfard, F. et al. Single-nucleotide-resolution computing and memory in living cells. Mol. Cell 75, 769–780.e764 (2019).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Zeng, H. et al. Recent advances in prime editing technologies and their promises for therapeutic applications. Curr. Opin. Biotechnol. 86, 103071 (2024).
Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015).
Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998).
Zhao, F. et al. Multiplex base-editing enables combinatorial epigenetic regulation for genome mining of fungal natural products. J. Am. Chem. Soc. 145, 413–421 (2023).
Glaser, A., McColl, B. & Vadolas, J. GFP to BFP conversion: a versatile assay for the quantification of CRISPR/Cas9-mediated genome editing. Mol. Ther. Nucleic Acids 5, e334 (2016).
Dingwall, C., Sharnick, S. V. & Laskey, R. A. A polypeptide domain that specifies migration of nucleoplasmin into the nucleus. Cell 30, 449–458 (1982).
Kalderon, D., Roberts, B. L., Richardson, W. D. & Smith, A. E. A short amino acid sequence able to specify nuclear location. Cell 39, 499–509 (1984).
Suzuki, K. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016).
Nair, R., Carter, P. & Rost, B. NLSdb: database of nuclear localization signals. Nucleic Acids Res. 31, 397–399 (2003).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
Das, D. & Georgiadis, M. M. The crystal structure of the monomeric reverse transcriptase from moloney murine leukemia virus. Structure 12, 819–829 (2004).
Zheng, C. et al. A flexible split prime editor using truncated reverse transcriptase improves dual-AAV delivery in mouse liver. Mol. Ther. 30, 1343–1351 (2022).
Gao, Z. et al. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol. Ther. 30, 2942–2951 (2022).
Grünewald, J. et al. Engineered CRISPR prime editors with compact, untethered reverse transcriptases. Nat. Biotechnol. 41, 337–343 (2023).
Böck, D. et al. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med. 14, eabl9238 (2022).
Oscorbin, I. P. & Filipenko, M. L. M-MuLV reverse transcriptase: Selected properties and improved mutants. Comput. Struct. Biotechnol. J. 19, 6315–6327 (2021).
Nowak, E. et al. Structural analysis of monomeric retroviral reverse transcriptase in complex with an RNA/DNA hybrid. Nucleic Acids Res. 41, 3874–3887 (2013).
Li, X. et al. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–34975 (1998).
Gilon, T., Chomsky, O. & Kulka, R. G. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 17, 2759–2766 (1998).
Pan, C. et al. Boosting plant genome editing with a versatile CRISPR-Combo system. Nat. Plants 8, 513–525 (2022).
Qi, LeiS. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Yeo, N. C. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Paddison, P. J., Caudy, A. A., Bernstein, E., Hannon, G. J. & Conklin, D. S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 16, 948–958 (2002).
Brummelkamp, T. R., Bernards, R. & Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553 (2002).
Moffat, J. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 (2006).
Elbashir, S. M., Martinez, J., Patkaniowska, A., Lendeckel, W. & Tuschl, T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 20, 6877–6888 (2001).
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652.e5629 (2021).
Chang, I. J., Hahn, S. H. Chapter 3—The genetics of Wilson disease. In Handbook of Clinical Neurology (eds Członkowska, A., & Schilsky, M. L.) (Elsevier, 2017).
Ferenci, P., Litwin, T., Seniow, J. & Czlonkowska, A. Encephalopathy in Wilson disease: copper toxicity or liver failure? J. Clin. Exp. Hepatol. 5, S88–S95 (2015).
Ferber, S. et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat. Med. 6, 568–572 (2000).
Liao, H.-K. et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171, 1495–1507.e1415 (2017).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018).
Hawkins, P. N. et al. Evolving landscape in the management of transthyretin amyloidosis. Ann. Med. 47, 625–638 (2015).
Abelson, J., Trotta, C. R. & Li, H. tRNA Splicing*. J. Biol. Chem. 273, 12685–12688 (1998).
Doman, J. L. et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell 186, 3983–4002.e3926 (2023).
Yan, J. et al. Improving prime editing with an endogenous small RNA-binding protein. Nature 628, 639–647 (2024).
Li, X. et al. Chromatin context-dependent regulation and epigenetic manipulation of prime editing. Cell 187, 2411–2427 (2024).
Park, S.-J. et al. Targeted mutagenesis in mouse cells and embryos using an enhanced prime editor. Genome Biol. 22, 170 (2021).
Zeisel, A., Yitzhaky, A., Bossel Ben-Moshe, N. & Domany, E. An accessible database for mouse and human whole transcriptome qPCR primers. Bioinformatics 29, 1355–1356 (2013).
Hsu, J. Y. et al. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 12, 1–6 (2021).
Fakhr, E., Zare, F. & Teimoori-Toolabi, L. Precise and efficient siRNA design: a key point in competent gene silencing. Cancer Gene Ther. 23, 73–82 (2016).
Schrödinger L. The PyMOL Molecular Graphics System, Version 2.1. (2015).
Acknowledgements
Q.Y. is a Sunit Patel ’85 Endowed Fellow supervised by X.G. This work was supported by the National Science Foundation CAREER AWARD (CBET-2143626) and National Institutes of Health grants (HL157714) to X.G.
Author information
Authors and Affiliations
Contributions
Q.C. Yuan, H.Z., and X.G. designed the research. Q.C. Yuan, H.Z., T.C.D., Q.L., Y.Y., E.C.O., Q.C. Yang, A.P., L.M.A., and B.Z. performed the experiments. Q.C. Yuan, H.Z., T.C.D., Q.L., Y.Y. analyzed data. Q.C. Yuan, H.Z., T.C.D., and X.G. wrote the manuscript with help from all authors. X.G. and X.Y. supervised the project.
Corresponding author
Ethics declarations
Competing interests
Q.C. Yuan, H.Z., T.C.D., and X.G. have submitted a provisional patent application to the U.S. Patent Office pertaining to the genetic perturbation technologies described within this work (application number 63/712,648). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yiping Qi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yuan, Q., Zeng, H., Daniel, T.C. et al. Orthogonal and multiplexable genetic perturbations with an engineered prime editor and a diverse RNA array. Nat Commun 15, 10868 (2024). https://doi.org/10.1038/s41467-024-55134-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55134-9
